 (FRA)
(FRA) 
Comprendre la Porphyrie Hépatique Aiguë
La Porphyrie Hépatique Aiguë (PHA) fait référence à une famille de maladies génétiques extrêmement rares, caractérisées par des crises menaçant potentiellement le pronostic vital et, pour certains patients, des manifestations chroniques et des complications à long terme qui ont des répercussions néfastes sur le fonctionnement au quotidien et la qualité de vie. Les PHA se partagent en quatre types, chacun d’eux étant associé à des anomalies distinctes d’une enzyme de la voie de biosynthèse de l’hème dans le foie : la Porphyrie Aiguë Intermittente (PAI), la Coproporphyrie Héréditaire (CH), la Porphyrie Variegata (PV) et la Porphyrie par déficit en ALA déshydratase (ADP). Les types de porphyries sont classés en deux catégories, les porphyries hépatiques aiguës et porphyries cutanées. Les porphyries cutanées sont des troubles métaboliques héréditaires rares qui se manifestent par la formation de vésicules sur la peau, des douleurs et/ou des rougeurs et un gonflement sur les zones exposées au soleil.
Aux États-Unis et en Europe, environ 5 000 personnes subissent une ou plusieurs crises de PHA chaque année, et environ 1 000 personnes souffrent de crises fréquentes et sévères.
Les symptômes de la Porphyrie Hépatique Aiguë peuvent avoir un impact sur la qualité de vie
Les personnes touchées peuvent présenter une combinaison des symptômes suivants :
Douleurs abdominales diffuses sévères, vomissements/nausées, urines foncées/rougeâtres

Faiblesse musculaire, engourdissement, insuffisance respiratoire

Confusion, anxiété, convulsions, hallucinations, fatigue

Lésions vésiculaires, érosions ou ulcérations de la peau exposée au soleil (en cas de PV et de CH)

Quelles sont les causes de la Porphyrie Hépatique Aiguë ?
Chez les personnes porteuses de l’anomalie génétique de la PHA,
l’une des enzymes impliquées dans la voie qui produit l’hème est
déficiente. Certains facteurs déclenchants peuvent avoir un impact
sur la voie et provoquer une augmentation de l’activité de l’ALAS1
(acide aminolévulinique synthase 1)
Cette augmentation de l’activité de l’ALAS1 se traduit par une accumulation d’intermédiaires neurotoxiques - l’acide aminolévulinique (ALA) et le porphobilinogène (PBG) - dans l’ensemble de l’organisme.
L’ALA et le PBG sont nocifs pour les cellules nerveuses et sont des facteurs associés aux crises et manifestations de la maladie caractéristiques de la PHA.
Quelles sont les causes de la porphyrie hépatique aiguë ?
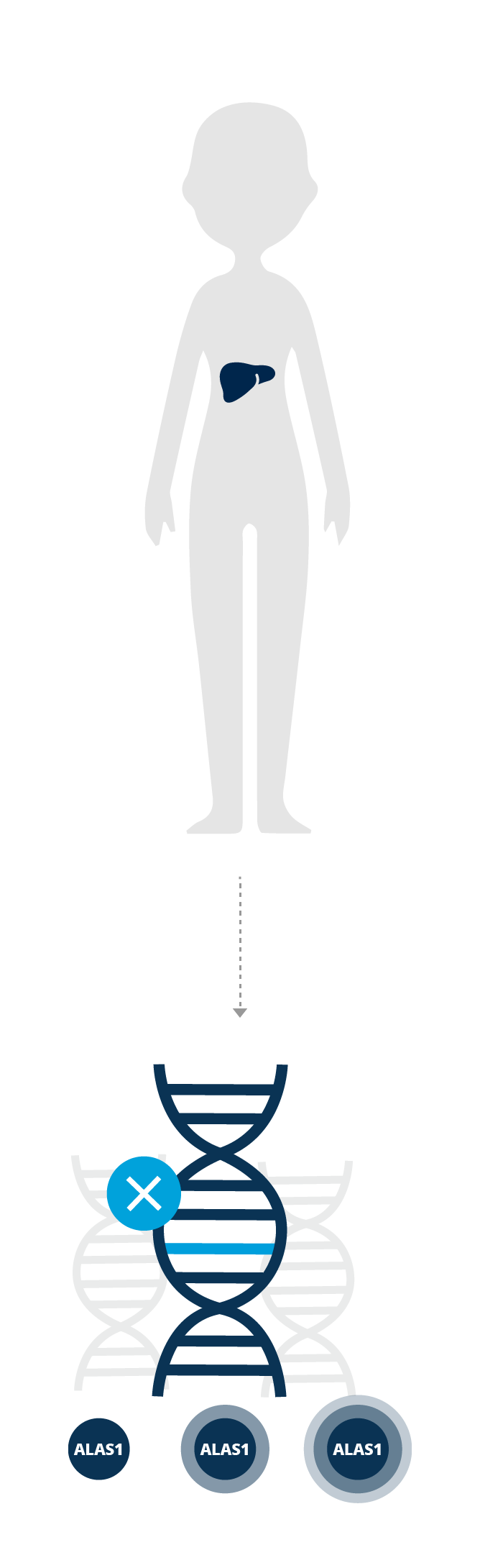
Chez les personnes porteuses de l’anomalie génétique de la PHA, l’une des enzymes impliquées dans la voie qui produit l’hème est déficiente. Certains facteurs déclenchants peuvent avoir un impact sur la voie et provoquer une augmentation de l’ALAS1 (acide aminolévulinique synthase 1).

Cette augmentation de l’ALAS1 se traduit par une accumulation d’intermédiaires neurotoxiques - l’acide aminolévulinique (ALA) et le porphobilinogène (PBG) - dans l’ensemble de l’organisme.

L’ALA et le PBG sont nocifs pour les cellules nerveuses et sont des facteurs associés aux crises et manifestations de la maladie caractéristiques de la PHA.

En savoir plus sur PHA



Ressources destinées aux professionnels de santé

Références
Puy, Hervé et al., Lancet 2010;375:924-937
ORPHANET; The Porphyria Consortium
Bissell DM et al., N Engl J Med 2017;377:862-872
Simon et al., Patient 2018;11:527–37
Pischik & Kauppinen. Appl Clin Genet 2015;8:201–14
Balwani & Desnick., Hematology Am Soc 6. Hematol Educ Program 2012;2012:19–27
Bonkovsky et al., Am J Med 2014;127:1233–41
Gouya et al., European Association for the Study of the Liver (EASL) Congress 2018. Presentation
Naik et al., Mol Genet Metab 2016;119:278–83
Szlendak et al., Adv Clin Exp Med 2016;25:361–8
Besur et al., Metabolites 2014;4:977–1006
Ramanujam & Anderson. Curr Protoc Hum Genet 2015;86:17.20.1–26
Elder G, et al., J Inherit Metab Dis 2013; 36(5) 849-57
Pallet N et al., Clin Kidney J 2018;11:191-197
Peoc’h K., Mol Genet Metabol 2018;10.001