



Nossas novidades
HYPEROXALURIE PRIMITIVE
Comprendre l’hyperoxalurie primitive de type 1
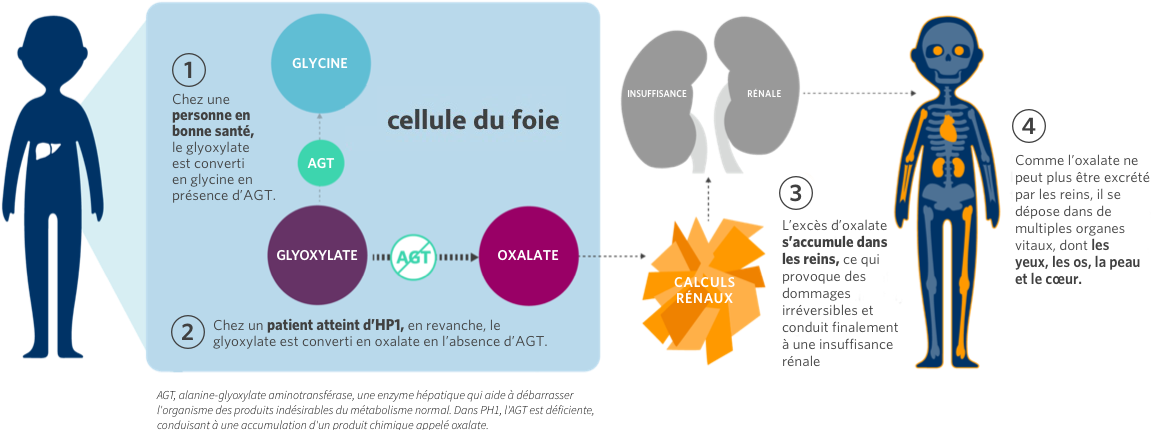
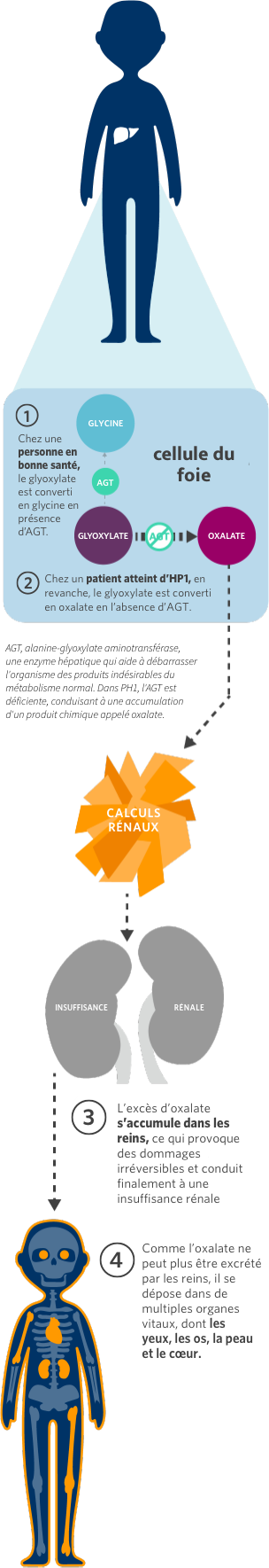
L’Hyperoxalurie Primitive (HP) constitue un groupe de maladies héréditaires rares du foie caractérisées par la surproduction d’oxalate, un produit final du métabolisme. Des niveaux élevés d’oxalate sont toxiques car l’oxalate ne peut pas être assimilé par le corps humain et s’accumule dans les reins.
Il existe 3 types d’HP : type 1 (HP1), type 2 (HP2) et type 3 (HP3). L’HP1 est la forme la plus courante et la plus grave, représentant 70 à 80 % des cas. L’HP1 est une maladie héréditaire rare dans laquelle des quantités excessives d’oxalate sont produites par le foie. L’HP1 touche environ 1 à 3 cas par million d’habitants.
La prévalence pourrait être sous-estimée puisque 5% à 10% des patients reçoivent un diagnostic d’hyperoxalurie primitive (HP) lors d’une récidive de la maladie après transplantation rénale. Dans certaines régions, telles que le Moyen-Orient et l’Afrique du Nord, la prévalence génétique de l’HP1 est plus élevée.
Quels sont les symptômes de l’HP1 ?
Les personnes atteintes d’HP1 sont souvent confrontées à la formation de calculs d’oxalate dans les voies urinaires et les reins. Lorsqu’une personne atteinte d’HP1 a un calcul rénal, les symptômes peuvent comprendre :
• Infections des voies urinaires
• Sang dans les urines
Certaines personnes ne sont diagnostiquées qu’après une défaillance rénale et nécessitent une dialyse pour aider à filtrer les déchets du sang.
L’HP1 dans l’organisme
Apparition et progression de la maladie


Les patients peuvent être diagnostiqués d’une HP1 à tout âge, mais la plupart des personnes ressentent leurs premiers symptômes dans la petite enfance.
Pour de nombreux patients, l’HP1 n’est pas diagnostiquée immédiatement. Étant donné que les calculs rénaux sont plus courants chez l’adulte, les patients adultes atteints d’HP1 passent souvent de nombreuses années sans être diagnostiqués, jusqu’à ce qu’ils présentent une maladie rénale avancée.
À mesure que l’HP1 progresse, elle se traduit souvent par une insuffisance rénale terminale, une affection qui met la vie en danger et qui empêche les reins de filtrer efficacement les fluides et les déchets de l’organisme.
Par conséquent, l’accumulation d’oxalate peut entraîner le dépôt de cristaux d’oxalate dans les yeux, les os, la peau, le cœur et le système nerveux central, provoquant une diminution de la vision, des fractures osseuses, des ulcères, une insuffisance cardiaque et d’autres complications.

Ressources destinées aux patients
Ressources destinées aux professionnels de santé

Références
Edvardsson VO, Goldfarb DS, Lieske JC, et al. Hereditary causes of kidney stones and chronic kidney disease. Peadiatr Nephrol. 2013;28:1923-1942.
Hopp K, Cogal A, Bergstralh E, et al. Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol. 2015;26:2559-2570.
Milliner DS, Harris PC, Cogal AG, Lieske JC. Primary hyperoxaluria type 1. GeneReviews, University of Washington, Seattle. 1993-2018.
Lorenz EC, Michet CJ, Milliner DS, Lieske JC. Update on oxalate crystal disease. Curr Rheumatol Rep. 2013;7:340.
Zhao F, Bergstralh EJ, Mehta RA, et al. Predictors of incident ESRD among patients with primary hyperoxaluria presenting prior to kidney failure. Clin J Am Soc Nephrol. 2016;11:119-126.
Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009;75:1264-1271.
Cochat P, Deloraine A, Rotily M, Olive F, Liponski I, Deries N. Epidemiology of primary hyperoxaluria type 1. Nephrol Dial Transplant. 1885;10(Suppl 8):3-7.
Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013,369:649-658.
Harambat J, Fargue S, Acquaviva C, et al. Genotype–phenotype correlation in primary hyperoxaluria type 1: the p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int. 2010;77:443–449.
RELATIONS CLIENTS - Pour toute question commerciale, une ouverture de compte ou le suivi de vos commandes :
serviceclients@alnylam.com | Tel : 01 87 65 09 22
Le contenu de ce site est destiné aux personnes résidant en France.